
Editor’s Note (10/8/21): David Julius, one of the authors of this article from 2006, is the co-recipient of the 2021 Nobel Prize in Physiology or Medicine for discoveries related to how the human body senses temperature and touch.
Throbbing, itching, aching, stabbing, stinging, pounding, piercing. Pain comes in a range of unpleasant flavors. But all pain has one thing in common: those who endure it want it to stop.
Yet the most widely used analgesics today are essentially folk remedies that have served for centuries: morphine and other opiates derive from the opium poppy, and aspirin comes from willow bark. Although these treatments can give relief, each has its limitations. Aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, cannot ease the most severe types of discomfort. And even opiates, generally the strongest medicines, do not work for everyone. Moreover, they can have serious side effects, and patients tend to become tolerant to them, requiring escalating doses to get any relief at all.
Over the past 20 years neurobiologists have learned a great deal about the cellular circuits and the specialized molecules that carry pain signals. Today this knowledge is being exploited to devise new strategies for managing pain better and causing fewer side effects. Indeed, more approaches than we have room to dicuss are now under study.
Particles of Fire
In the 17th century French philosopher René Descartes enumerated a theory to explain how people sense pain. In his view, a pinch, a whack or a poke essentially tugged on a neural rope that then rang a pain alarm bell in the brain. Imagine, for example, burning a foot. “Fast moving particles of fire,” Descartes thought, would create a disturbance that “passes along the nerve filament until it reaches the brain.”
Descartes was not too far off. Pain generally begins at the periphery: in the skin, an internal organ or any other site outside the central nervous system (CNS)—that is, outside the brain and spinal cord. Stubbing a toe or leaning against a hot stove activates neurons (nerve cells) called nociceptors that respond specifically to hurtful stimuli, such as extreme temperature or mechanical pressure, or to chemicals generated in response to injury or inflammation.
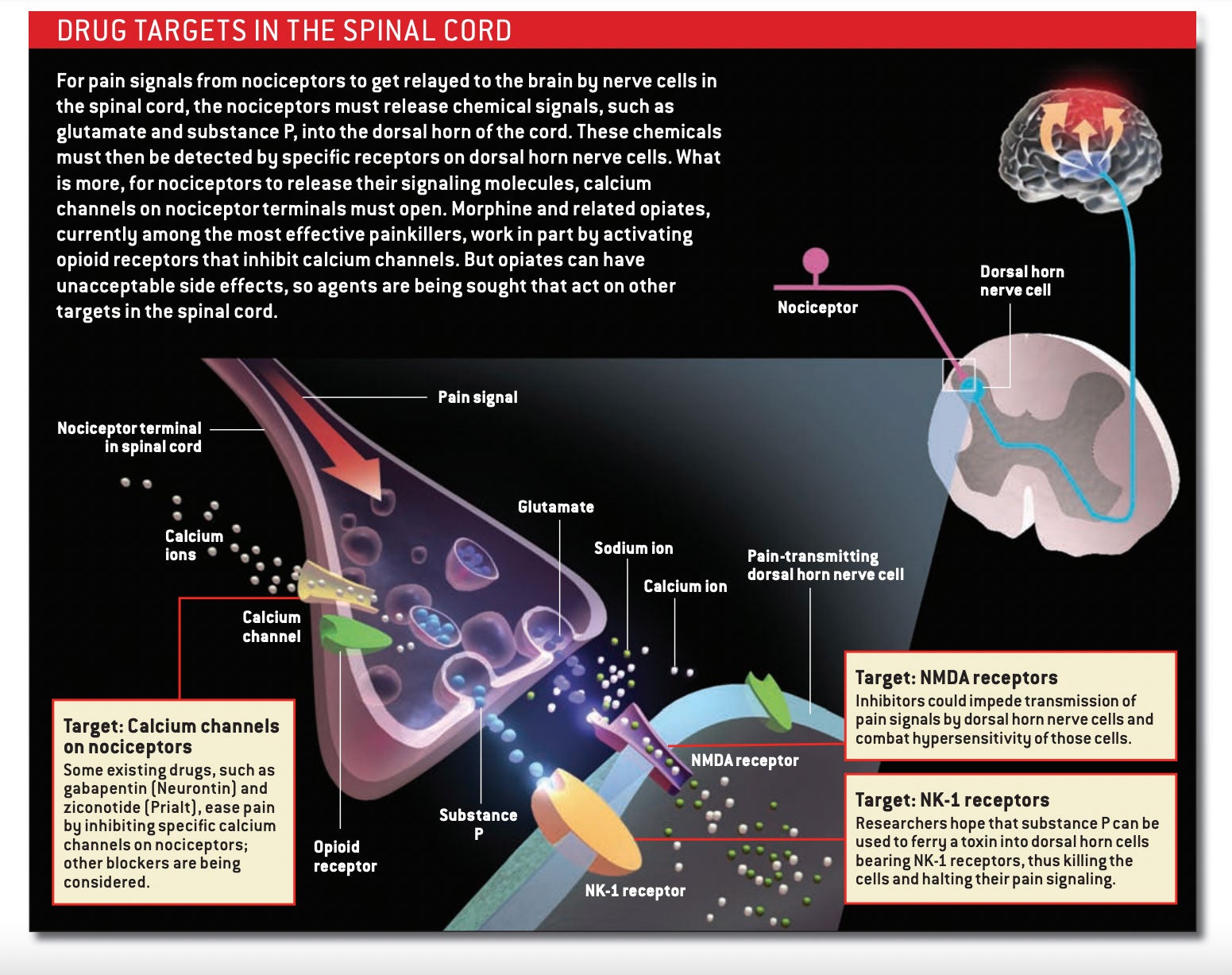
Nociceptors have two arms: a sensation-detecting branch that projects out to the periphery, where it innervates small patches of tissue, and a second branch that extends into the spinal cord [see box below]. The neuron’s cell body, which resides in a structure outside the spine, sits between the two. When specialized detector molecules on the peripheral branch encounter a noxious agent in the skin or an organ, they trigger an impulse that travels up the line, along the central branch and on to an area of the spinal cord known as the dorsal horn. There the nociceptor releases signaling molecules called neurotransmitters that activate neurons in the dorsal horn, prompting them to transmit the alarm message up to the brain. Although nociceptors are often called pain-sensing neurons, they merely indicate the presence of potentially harmful stimuli; it is the brain that interprets the signal as painful and prompts us to say “ouch.”
Not all pain is worrisome. For example, the acute kind that accompanies a minor tissue injury such as a sprain or abrasion is protective: it encourages an organism to avoid further injury. This kind tends to be temporary and to subside over time.
The pain that most troubles patients—and doctors—fails to disappear and is difficult to treat. In many cases, the problem arises because the injury or the inflammation that triggers the discomfort persists. The aches of arthritis result from ongoing inflammation, and the agony that can accompany invasive cancer stems to a large extent from tissue injury and inflammation.
In other cases, persistent pain is neuropathic, resulting from damage to nerve cells themselves. It can develop when neurons in the CNS sustain damage from multiple sclerosis, a stroke or spinal cord injury, for example. Or it can derive from injury to peripheral neurons. Amputees who endure aching in a limb that is no longer there (phantom limb pain) and people who feel burning in their skin for years after a herpes infection has subsided (postherpetic neuralgia) are all suffering from neuropathic pain. When this kind of hurt continues, it is not symptomatic of some ongoing injury or another disease; it is itself a disease of the nervous system and requires the attention of a pain specialist.
Pain without End
A major common denominator in those who suffer from hard-to-manage pain is abnormal sensitivity to stimuli. This sensitivity can take the form of hyperalgesia (an excessive reaction to typically painful inputs) or allodynia (pain in response to normally innocuous inputs). In those affected by allodynia, even the gentle pressure of clothing against one’s skin or bending a joint can become unbearable.
Biologists now understand that such heightened sensitivity—or sensitization— stems from molecular or structural changes in nerve cells. In the periphery, for instance, molecules that promote inflammation may cause the nociceptors that detect noxious stimuli to become overly reactive to those inputs. Inflammatory molecules can even cause nociceptors to begin generating signals in the absence of any environmental input.
Sensitization can also result from CNS changes that lead to hyperactivity of pain-transmission pathways. The changes, which may persist for a long time, can include display of increased numbers of the receptors that respond to the neurotransmitters released by nociceptors and might even include rewiring of connections and a loss of nerve cells that normally inhibit pain signaling. When the CNS is involved, the condition is termed central sensitization.
Regardless of which specific processes are at fault, ongoing pain, it turns out, can lead to sensitization and thus exacerbate and prolong discomfort. Many researchers, therefore, have amelioration of hyperalgesia and allodynia foremost in their minds as they hunt for new analgesics. Meanwhile patients need to realize that persistent pain should not be borne stoically; it requires aggressive treatment to prevent further sensitization.
Start at the Beginning
In the search for new analgesics, much effort has been directed toward the place where hurtful signals typically originate: the periphery. Certain of the specialized molecules that nociceptors use to detect noxious stimuli rarely occur elsewhere in the body. Blocking these molecules would presumably shut off pain signaling without disrupting other physiological processes and, thus, without causing unwanted side effects.

Credit: AMADEO BACHAR
Today’s most popular remedies—aspirin and other NSAIDs—largely work their magic in the periphery. When a tissue is injured, a variety of cells in the area pump out chemicals called prostaglandins, which act on the pain-sensing branches of nociceptors, lowering their activation threshold. Aspirin and NSAIDs inhibit the activity of a family of enzymes (cyclooxygenases) that cells use to generate the pain-inducing prostaglandins. These over-the-counter compounds relieve everyday aches and pains. But they also inhibit prostaglandin production elsewhere in the body, often causing such side effects as stomach pain, diarrhea and ulcers. These problems can prevent the drugs’ long-term use and limit the doses that can be given.
To reduce the gastrointestinal consequences, pharmaceutical companies developed a family of drugs that target the enzyme cyclooxygenase-2 (COX-2). Because COX-2 does not normally operate in the stomach or intestinal tract, blocking its activity should not cause the same disruptions as traditional NSAIDs do. Whether they are in fact gentler on the stomach remains to be established. In the meantime, the drugs have problems of their own. Rofecoxib (Vioxx), a COX-2 inhibitor that had been prescribed for relief of arthritis pain, was removed from the market when it was found to boost the risk of heart attack and stroke. Other COX-2 inhibitors are also being scrutinized for ill effects.
Send in the Salsa
Discovery of targets that reside almost exclusively on nociceptors provided an opportunity to develop drugs that act selectively to relieve pain. A particularly appealing one is the capsaicin receptor. This ion channel, present in the membrane of many nociceptors, responds not only to capsaicin, the pungent ingredient in chili peppers, but also to distressful heat and to protons (the hydrogen ions that make substances acidic); protons are unusually abundant in inflamed tissue. In the presence of these chemicals or of temperatures above 43 degrees Celsius, the channel allows sodium and calcium ions to flood into the nociceptor, stimulating it to generate a signal that translates into the burning sensation induced by heat, inflammation or spicy food.
Substances that inhibit the capsaicin receptors should therefore dampen inflammatory pain. Indeed, in laboratory animals, such “antagonists” have been able to relieve the very severe pain caused by the acidic environment around tumors that have metastasized to and damaged bone tissue. Today many pharmaceutical companies are competing to develop capsaicin receptor antagonists.
The possibilities for manipulating the receptor do not end there. Ironically, in some instances, purposely stimulating capsaicin receptors can alleviate pain. Topical creams containing capsaicin are being prescribed to relieve the itching, prickling and stinging sensations that can accompany postoperative wound healing or nerve impairments stemming from HIV infection, bouts of herpes and diabetes. Exactly how the ointments work is unclear, although small doses over time might ultimately make the receptor less responsive to the usual stimuli or might cause depletion of the neurotransmitters emitted by nociceptors.
Block Other Channels
A different kind of molecule found on the peripheral terminals of nociceptors is also attracting interest as a drug target. All neurons possess sodium channels that open in response to changes in the voltage across the nerve cell membrane, generating the impulses that relay messages from one neuron to the next. Local anesthetics that temporarily inactivate such voltage-gated sodium channels currently treat a variety of different pains, particularly those arising from a trip to the dentist. The problem, though, is that those anesthetics have to be applied at the site of the discomfort: disabling sodium channels throughout the nervous system could be fatal.
Pain-sensing neurons, however, possess a subclass of voltage-gated sodium channels, known as the TTX-resistant type, that do not occur in the CNS. Investigators therefore hope that drugs able to block this subclass could be administered systemically (throughout the body) without ill effects. Moreover, studies suggest that such agents could well dampen inappropriate hyperactivity by injured peripheral nerves and thus might relieve some neuropathic pain. Unfortunately, the pharmaceutical industry has so far been unable to successfully develop selective inhibitors for such channels, in part because they closely resemble TTX-sensitive sodium channels, which appear widely throughout the nervous system.

Credit: AMADEO BACHAR
The channels could perhaps be selectively removed, however, with a new technique called RNA interference. The method relies on introducing into an organism tiny molecules known as small AMADEO BACHAR interfering RNAs (siRNAs). These siRNAs prevent the production of an unwanted protein by inducing the degradation of the molecules (messenger RNAs) that direct the protein’s synthesis. The technique is being studied in humans for certain retinal conditions, but turning RNA interference into a pharmacological intervention for pain will be challenging. As is true of gene therapy, a virus will most likely be needed to deliver siRNA, and this aspect has raised safety concerns. Time will tell whether the approach will be practical as a pain therapy, but it remains an exciting possibility.
Suppose drug companies do develop a so-called magic bullet analgesic: a compound that specifically and effectively eliminates the activity of one of the pain-transducing molecules on nociceptors. Would this intervention provide relief from intractable pain? Maybe not, because closing off a single entrance to the pain pathway might not be enough.
Imagine, for example, a pharmaceutical that knocks out the receptor for bradykinin—a small protein, or peptide, that is produced during inflammation in the periphery. Bradykinin powerfully stimulates nociceptors, and an antagonist that blocks its receptors would certainly prevent those receptors from activating nociceptors. But it would not stop the neurons from recognizing and responding to other pain-inducing molecules generated by injury or inflammation—protons, prostaglandins, and a protein called nerve growth factor, for example. Similarly, hobbling only the capsaicin receptors might not mitigate all proton-mediated pain, because under certain circumstances, protons activate a separate population of detectors, called ASICs (acid-sensing ion channels), on nociceptors.
Focus on the Cord
One way around this redundancy problem would be to administer a cocktail of inhibitory molecules that targets multiple pain mechanisms. Another approach, though, would target molecules that act more centrally, blocking the ability of all nociceptors—no matter what stimuli initially activated them—to pass their pain signals to spinal cord neurons.
Morphine and other opiates, which bind to opioid receptors on the nociceptor endings that reach into the spinal cord, employ this latter tactic. In activating these receptors, opiates prevent neurotransmitter release, thus blocking the transmission of the pain message to spinal cord neurons. They also render dorsal horn neurons less able to respond to pain signals. Because these drugs act in the spinal cord, they should in theory be able to treat all types of pain, although they tend to work best against those related to inflammation.
Unfortunately, opioid receptors are present on neurons throughout the body, including in the brain and the gastrointestinal system. This ubiquity explains why morphine and its cousins can generate a broad set of undesirable side effects, including severe constipation and respiratory shutdown. These problems can restrict the amount of drug a patient can take safely or that a doctor will prescribe. And many physicians are reluctant to prescribe opiates for fear patients will become addicted. Addiction, however, is not common in those who take opiates only for pain. In part to avoid some of the undesirable effects, opiates are often delivered directly into the fluid-filled space around the spinal cord (intrathecally). The medicines may also be administered by injection (for postoperative pain) or via an indwelling pump (for chronic pain).
Alternatives to opiates are available as well. Medicines that interfere with calcium channels can prevent the release of neurotransmitters from nociceptor endings in the spinal cord. Gabapentin (Neurontin), an anticonvulsant, is believed to relieve some forms of pain by interacting with a specific subunit of certain calcium channels. And a relatively new drug called ziconotide (Prialt)—derived from the venom of a Pacific Ocean cone snail—inhibits a different variety of calcium channel known as the N-type.
Like opioid receptors, N-type calcium channels occur throughout the nervous system. If ziconotide were delivered systemically, blood pressure would decline precipitously. So the compound is administered intrathecally. Although the toxin blocks pain, its action within the CNS can still generate unpleasant side effects, including dizziness, nausea, headache and confusion. Ziconotide, therefore, is given mostly to patients with late-stage cancer who cannot get relief another way.
Recently drugs that act on cannabinoid receptors—the ones that mediate marijuana’s effects—have been advancing through clinical trials. These agents seem to ease pain in several ways, including by interfering with signal transmission between nociceptors and their target cells and by reducing the activity of inflammatory cells.
Batten Down the Hatches
Some investigators are concentrating on preventing spinal neurons from responding to neurotransmitters released by nociceptors—particularly to the amino acid glutamate, the primary carrier of the pain message. Glutamate activates various receptors in the dorsal horn of the spinal cord. Of these, the NMDA class participates in central sensitization, which makes it a logical target for new analgesics.
Credit: AMADEO BACHAR
Every neuron in the body possesses some type of NMDA receptor. Consequently, inhibiting all types at once would elicit catastrophic effects, including memory loss, seizures and paralysis. To avoid such reactions, researchers are attempting to hobble the receptor by acting on versions found mostly in the dorsal horn. Compounds that bind to a form containing what is called the NR2B subunit have yielded encouraging results in animal studies. For example, mice that had an NR2B inhibitor delivered directly into the spinal fluid were less sensitive to pain than were untreated animals. The drug also reversed allodynia in mice that had a peripheral nerve injury.
A number of nociceptors also release peptide neurotransmitters, such as substance P and calcitonin gene–related peptide (CGRP). These peptides activate pain-transmission neurons in the spinal cord by acting on discrete receptors, so drugs that bar interaction with those receptors would be expected to be helpful. Regrettably, selective blockade of the receptor used by substance P—the neurokinin-1, or NK-1, receptor—has failed in clinical trials for pain, perhaps because blocking that receptor by itself is insufficient. Whether quieting CGRP activity in the spinal cord will shut down pain is unknown, although the pharmaceutical industry is developing antagonists that aim to ease the agony of migraines by interfering with the release of CGRP onto blood vessels on the surface of the brain.
Kill the Messenger?
If all attempts to modulate pain signaling fail, one can consider killing the messenger. Cutting nociceptive nerves, though, generally backfires because, as we have noted, nerve injury can promote the onset of even more stubborn, persistent pain. Severing pathways in the spinal cord that convey information to the brain (cordotomy) was once common but now is reserved for terminal cancer patients unresponsive to all pain treatments. The problem with this last procedure is that the surgeon can not selectively cut the “pain” pathways.
A possible solution, now drawing considerable attention because of its success in animals, is a molecular therapy that takes out a subset of the spinal cord neurons receiving input from nociceptors. This cell-killing therapy couples saporin, a toxin, to substance P. The substance P in the conjugate binds to NK-1 receptors, leading to internalization of the whole compound, after which the saporin is freed to kill the neuron. Because the conjugate can enter only cells having an NK-1 receptor, researchers hope that side effects will be limited.

SOURCE: FRANZ F. HEFTI Rinat Neuroscience Corporation
Ablation of neurons in the spinal cord, however, should be considered a method of last resort: neurons in the CNS do not grow back, so the resulting changes—for better or worse—will be permanent. The same permanence does not hold in the peripheral nervous system, where cut fibers can regenerate. Ideally, therapies that trim back the signal-detecting branches of nociceptors—such as high doses of capsaicin—would halt pain but allow the branches to grow back eventually, restoring normal pain detection to the patch of tissue innervated before.
Targeting neurons may not be the sole way to overcome pain. Studies indicate that glia, the cells that nurture neurons in the CNS, swing into action in response to damage to peripheral nerves. The glia migrate to the region of the dorsal horn associated with the injured nerves. Then the glia discharge a bevy of chemicals that prod nociceptor terminals to release neurotransmitters in the cord, thus perpetuating a pain signal. Some of these substances, including growth factors and molecules known as cytokines, also make dorsal horn neurons overly excitable, and drugs blocking that hyperactivity should help undercut excessive sensitivity. Various groups are working to identify—and find ways to inhibit—the molecules that recruit and activate glia when nerves are damaged.
Interestingly, prostaglandins are among the key substances released from activated glia in the spinal cord. There they enhance pain by blocking receptors for glycine on dorsal horn neurons. Glycine, an inhibitory neurotransmitter, normally quiets these neurons. NSAIDs may therefore work not only by interfering with the production of prostaglandins in the periphery (the familiar way) but also by inhibiting COX enzymes in glia. In that case, direct delivery of COX inhibitors into the spinal fluid might minimize the side effects caused by systemic delivery. A pharmaceutical that enhanced glycine receptor activity could also help tamp the transmission of pain messages to the brain.
A Question of Perception
In this article we have discussed a subset of the experimental approaches to treating pain, all of which have shown promise in animal studies. Those evoking the greatest excitement leave normal sensation intact while diminishing the heightened sensitization characteristic of the difficult-to-treat inflammatory and neuropathic pains and have an acceptable side-effects profile. But will these therapies help patients? And will they work on all types of pain? These questions remain unanswered.
One approach that deserves further exploration is the use of behavioral, nondrug therapies for intractable pain—particularly that associated with conditions such as fibromyalgia and irritable bowel syndrome, for which no one has conclusively established an organic cause. Roughly a decade ago researchers at McGill University demonstrated that hypnosis could alter brain activity along with a person’s perception of pain. The scientists hypnotized volunteers and suggested to them that the hot water bath in which they had immersed their hands was either more unpleasant or less unpleasant than it really was.
Using positron-emission tomographic scanning to monitor brain activity, the investigators found that the somatosensory cortex, which responds to the magnitude of the physical stimulus, was equally active in both situations. But a second brain region, the cingulate cortex, was more active when subjects believed that the stimulus was more unpleasant, suggesting that hypnosis changed the way these individuals perceived sensations. By learning more about how the brain modulates the pain experience, investigators might be able to develop better cognitive therapies for moderating pain perception.
Poet Emily Dickinson often contemplated pain. In one work, she noted:
Pain has an element of blank;
It cannot recollect
When it began, or if there were
A day when it was not.
It has no future but itself.
We can only hope continued research into the mechanisms of pain sensation will lead to safe, effective treatments that will alter pain’s future, such that it reverts to a time when it was not.
