
For people living with rare genetic diseases—many of which can be debilitating or life-threatening—the need for innovative treatments is urgent. Only 5% of the 7,000 known rare diseases have an approved treatment, making patients with rare diseases collectively one of the most underserved communities in medicine today. Ultimately, the ability to realize the full potential of gene therapy to treat rare genetic diseases depends on listening to and addressing patients’ needs. Pfizer aims to establish a new paradigm; leading the way to bring new medicines to patients with a rare disease by including the patient voice at every stage of innovation and by leveraging the company’s expertise in rare disease research to develop a portfolio of potentially transformative recombinant adeno-associated virus (rAAV)-based gene therapies.
THE COLLECTIVE GLOBAL IMPACT OF RARE DISEASE
About 80% of rare diseases, defined in the United States as those that affect fewer than 200,000 people and in the European Union as diseases that affect no more than 1 in 2,000 individuals, are due to genetic mutations. Roughly half of all rare diseases predominately affect infants and children and can lead to significant illness and early death. While each genetic disease may occur in a small population of patients, collectively rare diseases affect approximately 400 million people worldwide and are responsible for significant loss of health, life and economic potential. The few treatments that have been available typically have focused on disease symptoms, without correcting the underlying cause of disease.
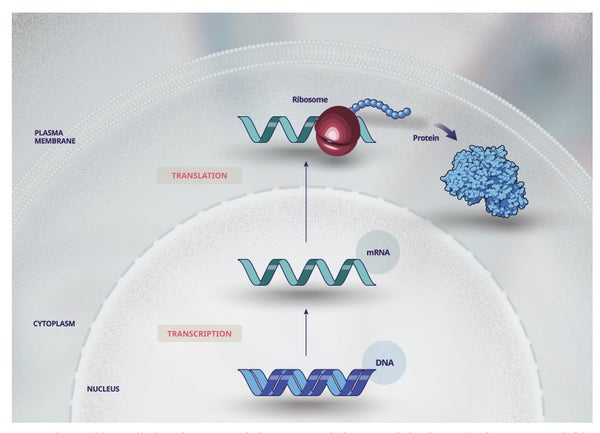
The symptoms and health impact of rare genetic diseases vary, but most of them result in the loss or alteration of a protein that is needed for cells and organs to function normally. Our genes are encoded in the DNA (deoxyribonucleic acid) of our chromosomes, which reside within the nucleus of the cell (Fig. 1) and contain the instructions to make all the proteins that our bodies need to be healthy. DNA is copied (transcribed) into RNA (ribonucleic acid), which leaves the nucleus and delivers these instructions to the protein making machinery (ribosomes) outside the nucleus.
Genetic disorders result from inherited or spontaneous changes in the DNA code (mutations). These changes can lead to the loss of function or gain in toxic function of a protein and subsequent alteration of cell function. Genetic medicines that restore functional patterns of protein expression hold great promise in treating rare diseases. Treatments include gene therapy (which the American Society of Gene and Cell Therapy defines as the introduction, removal or change in genetic material—specifically DNA or RNA—into the cells of a patient to treat a specific disease, although other sources may use different definitions), gene editing (permanently correcting, stopping or altering the mutation within the patient’s own DNA), and gene regulation (altering the transcription of the gene to RNA or translation of RNA to protein).
GENE THERAPY HAS THE POTENTIAL TO ADDRESS THE UNDERLYING CAUSE OF RARE DISEASES
Gene therapy represents the next step in the evolution of therapeutic development, which began with chemical compounds to treat symptoms (for example, aspirin to treat pain) and then advanced to biologic therapies (which could include proteins and antibodies) that modify disease (for example, enzyme replacement therapy to treat some inborn errors of metabolism, such as Gaucher disease). Now, gene therapy offers the potential to address the underlying biology of rare genetic diseases, which could reduce ongoing need for treatment, thus decreasing the treatment burden for patients and reducing health system demands. It may also improve quality of life for caregivers, especially parents, who today face significant emotional, physical and financial challenges. Enabling patients to live longer, healthier and more productive lives may further increase their ability to make positive contributions to their societies and economies.

Figure 2. From gene to protein using in vivo rAAV gene therapy. The rep and cap genes are removed from the wildtype (WT) adeno-associated virus (AAV) genome and replaced with an expression cassette that includes the therapeutic transgene, the promoter sequences needed to turn the gene on in targeted cell types, and a Poly A sequence that confers mRNA stability. Gene therapy for Duchenne muscular dystrophy and haemophilia B use transgenes encoding mini-dystrophin and factor IX, respectively. These therapeutic DNAs are then combined with an rAAV capsid to create an rAAV vector that is delivered to the patient by infusion or injection, depending on the target tissue. ITR - inverted terminal repeat of the wildtype AAV genome, which allows transgene replication during the manufacturing process.
INNOVATION IS ESSENTIAL FOR REALIZING THE PROMISE OF GENE THERAPY
Our bodies have multiple systems in place to keep foreign substances and DNA out of our cells. Safely and effectively transferring healthy genes to cells (Fig. 2) requires delivery methods that can bypass these systems and enable expression of the healthy gene for long periods of time in the specific cells affected by the disease.
Turning a harmless virus into a robust vehicle for gene therapy
Adeno-associated viruses (AAV) can be engineered so that the virus’ own genome is replaced with DNA that contains the functioning copy of the gene (known as a transgene) and a switch to turn the gene on in specific types of cells needed to treat a particular disease (known as a promoter). This combination of engineered DNA inside the AAV shell (the capsid) is called a vector (Fig. 2).
Engineered recombinant AAV vectors are the most common delivery system in current investigational gene therapy clinical trials, for several reasons. From a safety perspective, AAV is not known to cause disease in humans and it cannot make copies of itself without help from other viruses. This means that rAAV vectors cannot replicate in the human body. The healthy gene delivered by the rAAV vector also predominantly remains separate from the patient’s own DNA, which helps prevent generating additional mutations that might potentially occur if it were to be inserted into certain locations in the patient’s chromosomes. rAAV vectors have the potential to provide long-term expression of the healthy gene, and multiple strains of AAV (known as serotypes) enable development of rAAV vectors designed to preferentially target the cells needed to treat a particular disease (Fig. 2). Additionally, rAAV vectors can be delivered directly to the patient by infusion or injection (known as in vivo gene therapy). Importantly, rAAV vectors are already being utilized in gene therapies approved for use in the United States and Europe, demonstrating their viability. For all these reasons, Pfizer uses rAAV vectors for in vivo gene therapy as its main approach to rare disease gene therapy.

Figure 3. Pfizer's end-to-end, patient-centric approach to gene therapy development. Pfizer's approach to gene therapy spans discovery, development and delivery. Patient engagement by stage; Pfizer activity by stage; manufacturing and collaboration are critical end-to-end.
PFIZER’S END-TO-END rAAV GENE THERAPY PLATFORM
Pfizer has established what we believe to be an industry-leading end-to-end (discovery, through development and delivery) gene therapy platform to help realize the potential of gene therapy in treating a variety of rare genetic disorders and ensure that patients’ needs and perspectives are considered at every stage of development (Fig. 3). This end-to-end platform leverages Pfizer’s extensive rare disease drug development expertise to advance gene therapy development timelines, support global gene therapy trials, and engage early with patients to understand their needs. This approach has enabled one of the largest pipelines of Phase 3 gene therapy programmes in the industry (haemophilia A, haemophilia B, and Duchenne muscular dystrophy) and multiple preclinical programmes in rare haematology, neurology, cardiology and endocrine/ metabolic diseases.
Discover
In the discovery phase, Pfizer scientists evaluate the part that specific gene mutations play in disease. Then they determine the best way to deliver a healthy copy of the gene to the right cells in order to treat the disease. They also determine if there is an age or stage of disease before or after which gene therapy is not as likely to be effective. For example, Duchenne muscular dystrophy presents early, progresses rapidly and leads to muscle damage and failure over time. In this and other progressive diseases, gene therapy may prevent significant and irreversible damage when administered in childhood and may prevent further decline when administered to older patients.
A variety of activities happen during the development phase, including conducting clinical trials that prioritize the safety of participating patients and provide the safety and efficacy data that patients, physicians, regulatory authorities and other stakeholders need to make informed approval and treatment decisions. Several important considerations go into designing trials that achieve these goals. One consideration is determining which patients are most likely to benefit from the gene therapy being tested, which, as noted above, may depend on a number of factors. Another consideration is the approach to patients who may already have antibodies against AAV. Because AAV is a naturally occurring virus, many people have already been exposed to it, and their immune systems have been primed to prevent AAV (and related rAAV vectors) from entering cells. Some of these antibodies can keep rAAV vectors out of cells (known as neutralizing antibodies, or NAbs). Significant levels of NAbs may block the rAAV gene therapy from getting into enough cells to have a therapeutic effect. For this reason, many rAAV clinical trials currently exclude patients who have levels of NAbs above a certain value. From a safety standpoint, the immune response to AAV can potentially lead to adverse effects related to rAAV vectors. To reduce this risk, rAAV gene therapy clinical trials often include temporary use of medications that reduce the host immune response and have processes in place to monitor and respond to these events if they arise.
The assessment of whether a potential therapy provides benefit is determined by measuring changes in specific clinical trial assessments, known as endpoints. Endpoints can include changes in levels of the therapeutic protein produced by the delivered gene (for example, Factor VIII or Factor IX for haemophilia A and B, respectively, or mini-dystrophin for Duchenne muscular dystrophy), changes in physical functions such as walking speed or ability to climb stairs (Duchenne muscular dystrophy), bleeding rates in haemophilia and the patients’ own perspectives on how life with their disease has changed in response to the treatment. Choosing endpoints that matter to patients and that can be measured consistently and reproducibly may be challenging, especially for diseases in which consensus on how to define ‘benefit’ has not yet been reached. Therefore, consulting with patients, clinicians and regulators is essential for selecting endpoints that are meaningful to patients, can be used consistently in clinical practice, and provide regulators the information they need to evaluate and potentially approve a new therapy. Finally, creating registries that collect long-term data from patients who receive new gene therapies, once they are approved, is important for the continued evaluation of safety and efficacy over many years.
Develop
The development phase also includes working with regulators to establish a path towards potential product approval that balances the need for robust safety and efficacy data with the often-urgent unmet need for a new therapy. This is a complex process that necessitates effective collaboration among regulatory agencies, patients, patient advocacy groups, industry and academia. A comprehensive regulatory strategy also needs to reflect the priorities and processes of different global regulatory agencies. As a company with a legacy of more than 170 years of success in discovering, developing and delivering therapies to patients around the globe, Pfizer is leveraging its regulatory expertise to support its rare disease gene therapy pipeline. It is also engaged in collaborative efforts to harmonize regulatory requirements across different countries and regions to streamline the approval of rare disease gene therapies and ensure access to approved therapies as quickly and broadly as possible.
Deliver
The delivery phase comprises activities designed to ensure that approved gene therapies reach the patients who may benefit from them. This includes innovative work with payers and policy makers to develop novel solutions that ensure patient access to potentially one-and-done therapies that may provide benefit for many years or over a patient’s lifetime. Managing supply chain logistics and educating healthcare practitioners are critical for facilitating delivery of gene therapies to patients’ community care settings and sparing patients from the burden of travelling long distances to specialized treatment centres. Expanding access to genetic testing to enable early diagnosis of a rare genetic disease is also important for the delivery of gene therapies. This is especially true for rapidly progressive diseases in which early treatment may provide the greatest benefit.
Throughout discovery, development and delivery, Pfizer proactively engages with patients. During the discovery phase, patient insights illuminate scientists’ understanding about how rare genetic diseases impact their daily lives, what types of changes or improvements in their disease symptoms would be most meaningful to them, and what risks they are willing to accept or not accept to achieve those changes. Throughout clinical and regulatory development, patient input can help optimize clinical trial designs by identifying barriers to participation and identifying what measurements are considered meaningful and should be used to determine that a new therapy is effective. Patients also have an important part to play in ensuring that approved therapies can be readily delivered, by helping to educate regulators, policymakers, and payers about their needs and the potential value that a new therapy may provide to them and their caregivers.

Figure 4. Components of Pfizer's rAAV triple transfection manufacturing process. Pfizer's process to manufacture each of its rAAV gene therapy candidates requires a production cell line and three different components (known as triple transfection): the genetic material (transgene) to be carried by the vector, key replication and structural proteins for the vector (rep and cap), and other (helper) proteins that aid the replication and assembly of process. All three components are introduced into a production cell line (such as HEK293 or other established cell line). The transfected cells are cultured in bioreactors, where rAAV vector production takes place.
MANUFACTURING EXCELLENCE SUPPORTS SUCCESS
Clinical trials of investigational rare disease therapies often include smaller numbers of patients than those for larger disease indications, which may result in compressed clinical development timelines. Therefore, the manufacturing processes for these therapies need to be established and approved on an often shorter timeline than other types of therapies. This may lead to regulators having more questions about gene therapy manufacturing practices than with other types of drugs in development. The regulatory landscape for gene therapy manufacturing is also complicated by the fact that many aspects of the key regulatory requirements continue to be refined. Moreover, manufacturing requirements may differ among regulatory agencies, which adds complexity when trying to make these novel therapies, if approved, broadly available around the world.
Like the manufacture of other biologics, the first step in this process is developing what is known as a production cell line (Fig. 4). The production cells act as mini factories that make the rAAV vector. Once the production cell line has been created, the cells are grown in culture and transfected to produce trillions of rAAV vectors, depending on the disease indication. The number of rAAV vectors needed to treat a tissue such as muscle, which is located throughout the body, is tens of thousands of times greater than the number needed to treat, for example, retinal diseases for which the rAAV vectors can be injected directly into the retina.
Pfizer uses large vessels (also known as bioreactors, Fig. 4) like those used to manufacture the company’s other biologic therapies. The production cells must be lysed to release rAAV vectors into the growth medium in which the cells are growing. The liquid is collected and treated in several steps to remove any cellular debris or other manufacturing byproducts or impurities. Additional steps are also used to reduce the number of ‘empty’ vectors that do not contain the healthy gene. These multiple purification steps, each of which includes quality control tests, result in a high-quality rAAV vector product, which is then packaged for cold shipment and delivery. Manufacturing demands increase as a gene therapy candidate moves from the discovery stage through clinical trials, and approved gene therapy products which will require sustained production capable of meeting global patient demand.
The expertise in packaging and cold supply chain management that Pfizer developed in support of its gene therapy pipeline played a critical role in its ability to rapidly develop and deploy a cold chain process for delivering the Pfizer-BioNTech COVID-19 vaccine around the world. Additional insights gained from the deployment of the vaccine are being incorporated into innovative cold chain management processes for its gene therapy candidates. Processes that go beyond approaches traditionally used for biologic therapies are expected to enable more efficient delivery of rAAV gene therapies, if approved.
Pfizer’s end-to-end rAAV manufacturing process builds on the company’s expertise in manufacturing other biologic therapies, to produce high quality vectors that can be scaled to meet development stage and dosing requirements of its gene therapy candidates. This expertise is critical for preventing regulatory delays as gene therapy candidates move from clinical trial development to global delivery, and across a wide range of doses. The company has established a diversified manufacturing infrastructure to support end-to-end development through future delivery of its gene therapy candidates, which includes cutting-edge manufacturing facilities in Morrisville, Durham, and Sanford North Carolina in the United States. Combined, these facilities provide Good Laboratory Practice grade material at small scales (10-250 litres) for research and development activities and Good Manufacturing Practice grade material at a variety of scales for early (500 litres) and pivotal clinical (2,000 litres) trials and to ultimately meet global patient demand. The number of doses produced at each scale depends on the disease indication.
PFIZER’S PATH TO REALIZING THE POTENTIAL OF rAAV GENE THERAPY FOR RARE DISEASES
Pfizer is engaged in multiple internal and collaborative efforts to support the advancement of gene therapies.
Advancing science
Scientists are innovating novel rAAV vectors that may more effectively enter target cells and/or make larger amounts of the healthy protein these vectors encode. Researchers are also exploring multiple approaches for reducing potential immune responses to rAAV vectors, which is important for improving safety and efficacy when patients receive a first dose, and for potentially enabling patients to receive additional doses in the future if needed. One of these approaches is plasmapheresis, in which the patient’s blood is processed through an external device to remove NAbs prior to the administration of rAAV gene therapy. This is expected to prevent the immune system from blocking entry of the rAAV vectors into target cells. Another approach is to administer medications that temporarily block the immune system from making new antibodies against rAAV vectors. This could also increase the number of rAAV vectors that reach their target cells during initial dosing while blocking the development of antibodies that might reduce the efficiency of future doses, if needed. The temporary use of medications currently used to reduce the levels of antibodies, including NAbs, in some transplant patients may also provide benefit in reducing antibody responses to rAAV vectors in gene therapy patients.
The availability of these interventions may allow for inclusion of patients who could otherwise be excluded from gene therapy because of high levels of pre-existing NAbs to the AAV vectors. Moreover, while data from approved and investigational rAAV gene therapies demonstrate promising durability of therapeutic protein expression and thereby the potential as one-time therapies, second or even third doses may theoretically be needed in some diseases or for some patients. Pfizer is exploring multiple ways, including the use of combination interventions, to reduce immune responses to rAAV vectors, which is essential for supporting long-term expression from initial doses and enabling re-dosing if necessary.
Expanding global regulatory capacity
The current pace of gene therapy approvals is slower than expected, in many cases due to regulatory complexities. Reducing regulatory delays requires the development and implementation of consistent and standard guidance on clinical trial design and endpoints and clarity for guidance on the chemistry, manufacturing and controls used to produce gene therapies. Ensuring that new gene therapies are available to patients around the world also requires global integration and harmonization of regulatory guidance, and early and productive communication between regulators and gene therapy researchers. Pfizer is leveraging its global regulatory expertise and collaborating with scientific organizations, industry, regulators and others around the world to address the regulatory challenges facing gene therapy.
Developing innovative access models
Traditional therapies for rare genetic diseases are used throughout a patient’s life, and current reimbursement policies and payment models are not designed to accommodate gene therapies that may be dosed only once or a few times. Pfizer is working with governments and payers to develop innovative policies and access models that recognize the potential health, economic and societal benefits that gene therapies can provide over a patient’s lifetime, and to reduce financial and other barriers to accessing potentially transformative therapy.
The potential of rAAV vectors in enabling new transformational therapies for patients living with rare diseases has been validated. Fully realizing that potential requires continued advancement and innovation across the entire discovery, development and delivery continuum among all stakeholders.
For more information on Pfizer’s rare disease therapy portfolio, please visit https://www.pfizer.com/science/rarediseases/; information on Pfizer’s gene therapy programmes is available at https://www.pfizer.com/science/researchdevelopment/ gene-therapy.
