


Gene therapy is a promising approach to altering the genetic composition of cells as a way to correct disease-causing mutations or to express proteins or RNA molecules that confer a therapeutic benefit. The concept of gene therapy is straightforward: deliver nucleic acids to target cells to alter their function in a beneficial manner. Moving from concept to reality, however, is a complex process comprised of multiple steps and components, including systems for getting nucleic acids into target cells, DNA regulatory elements that control the amount, location and duration of gene expression, and production of proteins with appropriate activity to alter cellular function in the desired manner. Pfizer is currently focusing on diseases that have single-gene defects, such as certain neuromuscular and hematologic diseases, and we have a robust pipeline of potential gene therapy treatments in preclinical and clinical development. Our current portfolio includes a phase 1b clinical trial for Duchenne muscular dystrophy (DMD), a pivotal phase 3 program in hemophilia B as part of a collaboration with Spark Therapeutics, and an ongoing phase 1/2 trial in hemophilia A in collaboration with Sangamo Therapeutics, Inc.
Given the multiple elements required for successful gene therapy development, collaboration among industry, academia, regulatory, clinician and patient communities is essential. Such partnerships ensure that the necessary expertise is harnessed to achieve maximum benefit: gene therapy products that are clinically beneficial, meaningful to patients and commercially accessible.
Although the clinical utility of ex vivo gene therapy has been validated with multiple approved products (for example, Strimvelis, KYMRIAH and YESCARTA), the delivery of genes to cells in vivo has been more challenging. One factor that contributes to the challenge is the immune responses that patients may have to viral vectors or to transgene products that were not previously present in the patient’s body. Another challenge lies in the limited access that gene therapies may have to the surface of target cells in vivo as compared with cultured cells. As a result, gene therapy clinical trials conducted by other pharmaceutical companies over the past 20 years in diverse indications including cystic fibrosis and age-related macular degeneration have failed to advance successfully through clinical drug development.
However, advances in vector engineering, transgene optimization and the combinatorial use of regulatory elements over the past several years have addressed some of the challenges of in vivo gene therapy, and Pfizer believes that gene therapy for single-gene disorders is at a pivotal period in its evolution. In some ways, the field is following a trajectory similar to the development of biologic therapies, which have become common, and critical components of treatment regimens for many serious medical conditions, including cancers, neurologic disorders, diabetes and autoimmune diseases. Innovations in vector design and an enhanced understanding of the biology of certain diseases have enabled the development of novel gene therapy approaches, and numerous late-stage trials of next-generation product candidates, including from Pfizer, are ongoing. Data from these trials will hopefully advance our current understanding of the potential for gene therapy as a therapeutic option, while laying the foundation for future improvements.
The unmet needs of patients with single-gene disorders fall largely in two domains. One area of unmet need is found among patients with diseases for which therapy exists but may only be palliative or associated with high treatment burdens. For example, individuals with hemophilia B are able to address bleeding episodes through frequent intravenous blood transfusions and may be able to achieve disease management with recombinant Factor IX protein therapy. However, gene therapy may improve the quality of life for people with hemophilia B by reducing or eliminating the need for frequent dosing and by ensuring a stable level of Factor IX expression, thus avoiding the peaks and troughs associated with intravenous administration of Factor IX protein. The other type of unmet need is found among individuals with rare, single-gene disorders for which no treatments exist for the majority of patients. Examples in this category include debilitating, progressive disorders, such as DMD, spinal muscular atrophy and Friedreich’s ataxia.
The recent technological advances made in gene therapy have opened up the potential to address a broad array of challenges, and Pfizer is pursuing potential solutions in both areas of unmet need. However, achieving the goal of such gene therapies requires not only novel technology, but innovative approaches to solving the technical challenges inherent to the research and development process: translational science, robust and scalable manufacturing, and a collaborative model that takes into account the need to move forward all of these components in tandem. Herein we present Pfizer’s perspective on some of the ways in which companies can organize and collaborate in order to work towards realizing the full promise of gene therapy.
Translating disease biology to therapeutic strategies
Focus on understanding disease biology
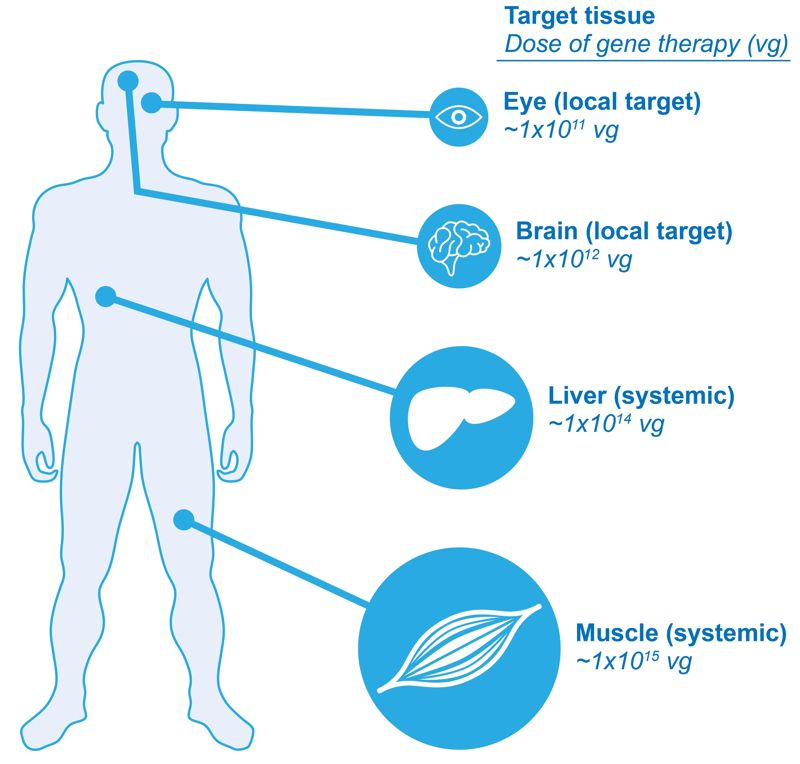
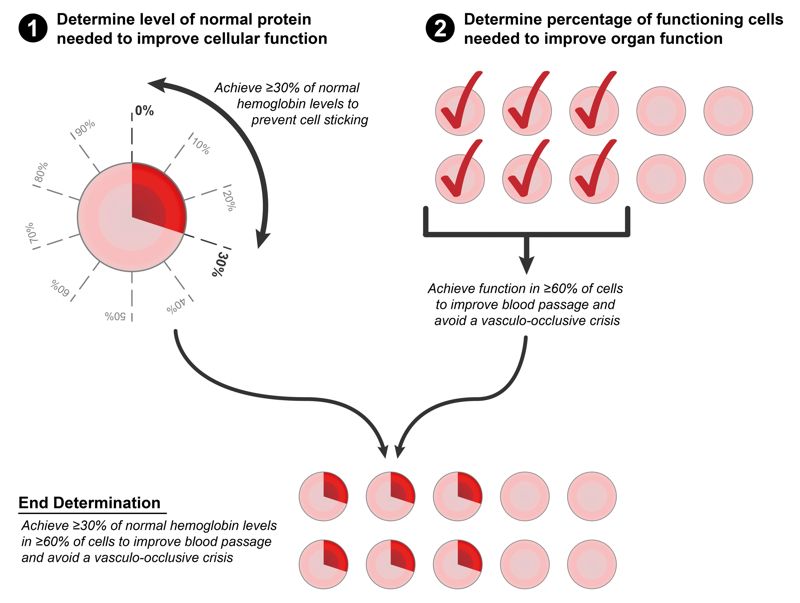
Although most conditions targeted by gene therapy result from alterations in the activity of a single gene, the effects of these alterations can vary among different cell types and tissues and at various points in a patient’s age or developmental stage. Consequently, safe and effective development of gene therapies requires clear insights into disease biology across various cells, tissues and patient demographics. This includes understanding which cell types and tissues need to be targeted and determining if those targets change over time or as a factor of disease progression. While target cell types that undergo frequent cell division require integrating vectors or gene editing strategies for effective treatment, non-dividing target cells can be treated with vectors that remain episomal. Establishing benchmarks for expression of the delivered gene, with respect to both the number of expressing cells and the overall level of expression (Fig. 1), is important for informing decisions about optimizing transduction and designing expression cassettes that confer clinically therapeutic levels and localization of the expressed protein. Some diseases, such as DMD, require protein expression predominantly within specific cell types (for example, skeletal and heart muscle). Other diseases, such as hemophilia, may have less stringent requirements for where the protein is expressed as long as it is secreted and at a level that restores correct biologic activity. Access to the tissues to be targeted in a specific disease also plays an important part in vector selection and design, and dosing strategies (Fig. 2). For example, in many ongoing clinical studies that use an adeno-associated virus (AAV) vector to deliver a gene, localized injection of AAV is being used as a dosing strategy to address tissue accessibility.
Hemophilia and DMD, two indications for which Pfizer has potential therapeutics being evaluated in the clinic, provide examples of the role that disease biology plays in gene therapy development. Hemophilia is an indication with a relatively wide therapeutic window with respect to expression levels and relatively straightforward tissue targeting requirements. This is because delivery of Factor VIII (for hemophilia A) or Factor IX (for hemophilia B) coding sequences to liver cells is expected to sufficiently restore blood clotting activity to significantly reduce the bleeding episodes seen in hemophilia. On the other end of the therapeutic range, healthy individuals show levels of Factor VIII or IX up to 150%1, thus overexpression of the clotting factor is not a concern. The ability to detect Factor VIII or IX activity in the plasma also provides an easily measurable biomarker that is directly related to disease severity and treatment effect. In contrast, DMD represents an indication with additional challenges in effectively transducing a sufficient number of muscle cells and achieving high enough levels of protein expression within those cells to achieve clinically relevant benefit. This is due to the large mass and broad distribution of the muscle tissues that are affected by the disease. The difficulty in accessing muscle cell samples and measuring intracellular dystrophin also complicates obtaining a biomarker of effect.
Freidreich’s ataxia exemplifies how disease biology may complicate therapy for a single disease because it affects both the central nervous system and cardiac function. Clinically relevant transduction rates and expression levels may vary among different tissue types and it may be challenging to develop a single gene therapy, or routes of administration, optimized for multiple anatomic compartments.
Optimizing vector design and engineering
Given the prominent role that disease biology plays in defining the requirements for potential gene therapies in specific diseases, it is unlikely that a single vector or expression system will be applicable to all indications that could be treated with this therapeutic modality. Thus Pfizer scientists believe it will likely be important to develop a suite of recombinant adeno-associated virus (rAAV)-based vector systems that can be deployed based on the desired product profile. AAV is one of the most actively employed vectors for in vivo gene therapies because of its versatility in targeted applications and its safety profile compared to other viruses. Compared to the wild-type AAV, which consists of a capsid shell encasing a single-stranded DNA, the rAAV used in gene therapy lacks components of its own viral DNA but retains the ability to deliver exogenous DNA (that is, an engineered transgene expression cassette) for therapeutic purposes into the nucleus of target cells. Capsid and transgene cassette engineering will therefore play a vital part in developing gene therapies that are optimized for safety and efficacy within specific indications (Fig. 3).
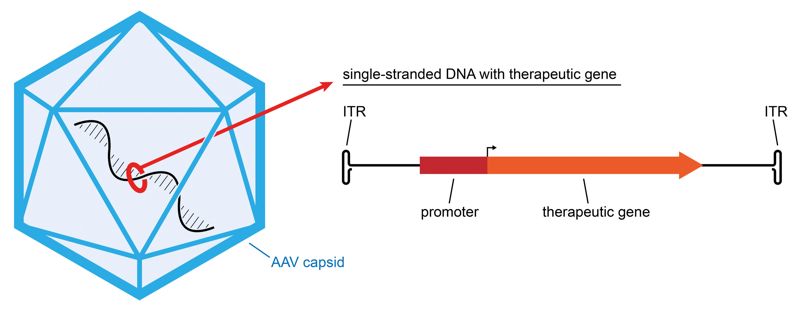
Capsid engineering has been used to improve delivery to and transduction of specific tissues, which is necessary for clinically meaningful protein expression. Naturally occurring AAV variants have different tissue tropisms owing to subtle differences in binding preferences of cell surface receptors, and those variants can be of use in targeting specific tissues. Additionally, engineering capsids to develop novel AAV variants, such as through peptide ligand insertion or directed evolution through capsid shuffling, may have further benefits for cell or tissue specificity.
Capsid engineering may also be employed to reduce anti-capsid immune responses that can interfere with transduction activity. AAV is less immunogenic compared with other viruses, primarily because it is unable to replicate or infect without the presence of a helper virus (such as an adenovirus). However, many people have already been exposed to some variant of AAV and may have a pre-existing adaptive immune response to that variant. The concern with such pre-existing immunogenicity is that circulating neutralizing antibodies (NAbs) and T cells may reduce clinical efficacy of AAV vectors. Approaches to overcoming this challenge may include selecting a vector that either has not previously circulated or that does not elicit a clinically significant adaptive immune response, as well as engineering AAV to modify particular antigenic domains on the capsid shell. In any of these cases, performing neutralization assays with living cells to quantify the expected immune response of a given capsid will be helpful in the selection of the vector.
Because a single-stranded transgene delivered to the nucleus of the target cell(s) needs to be converted to a double-stranded molecule to be expressed, this can be a rate limiting step in gene expression2,3. One established approach to overcoming this is to develop a self-complementary transgene to bypass the need for single- to double-stranded conversion. An example of such so-called transgene-cassette engineering is the approach that AveXis (now Novartis) took with the human SMN transgene for the treatment of spinal muscular atrophy (SMA), the leading genetic disease associated with infant mortality (www.avexis. com)4. This transgene is under the transcriptional control of the cytomegalovirus enhanced chicken beta-actin hybrid (CB) promoter (NCT03306277)4. A pivotal phase 3 trial of this optimized sequence delivered with an AAV9 vector, the same vector Pfizer is using in its DMD clinical trial, is currently ongoing (www.avexis.com; NCT03306277)4.
The use of naturally occurring gene variants offers another approach to optimizing protein activity5. For example, Spark and Pfizer’s fidanacogene elaparvovec comprises a naturally occurring variant of Factor IX that has been shown to increase clotting activity eight-fold compared with wild-type Factor IX5. Spark has further optimized the codon usage within this Factor IX variant to increase the expressed protein’s stability and activity and to reduce its immunogenicity (www.sparktx.com)6.
Another key component of vector engineering is the development of expression constructs that can fit within the size constraints of particular vector systems. For example, the payload capacity of most AAV vectors is less than 5 kb of sequence, which must comprise coding sequences as well as regulatory elements. DMD results from a loss of functional dystrophin protein. The DMD gene is one of the largest human genes, comprising 2.3 mb of DNA. The full dystrophin cDNA is 14 kb long and cannot be accommodated within an AAV vector. PF-06939926, Pfizer’s phase 1 investigational candidate for the treatment of DMD, utilizes a shortened version of the human dystrophin cDNA that has been engineered to provide essential protein function under the control of a human muscle-specific promoter that comprises less than 5 kb of DNA sequence (mini-dystrophin). Similarly, full-length cDNA for Factor VIII is approximately 7 kb of sequence, therefore a fully functional B-domain deleted version that can fit into AAV is being used in all current gene therapy trials.
Robust, scalable and reproducible manufacturing
At Pfizer, we believe that transforming gene therapy from a promising approach to a commercially viable therapeutic modality requires the development of robust, scalable and reproducible manufacturing processes. Consequently, the feasibility of commercial-scale manufacturing of a particular gene therapy candidate must be evaluated at the earliest stages of the development pathway. Manufacturing processes that are being developed by Pfizer also need to balance the goal of establishing a consistent manufacturing approach with the unique vector design requirements for individual disease indications.
One approach that we are exploring at Pfizer is to build internal manufacturing capabilities. This has the potential to provide Pfizer with direct control over process development and flexibility to implement process improvements and to address the parameters of particular gene therapy products.
Another key gene therapy manufacturing asset for Pfizer and other companies pursuing gene therapies is in-house plasmid production capabilities. The availability of high-quality plasmids can often be rate limiting for programs in development. The importance of plasmid quality is highlighted by the potential for the United States Food and Drug Administration (FDA) to place a clinical hold on a gene therapy trial if there is contamination of the plasmid used to manufacture clinical trial material.
Finally, development of robust, scalable manufacturing also requires constant innovation. The desire to innovate proprietary technology, which can be time and cost-intensive, needs to be considered alongside the importance of moving optimized new products toward patients as quickly as possible. Consequently, Pfizer’s strategy regarding a given manufacturing technology takes into account its potential clinical value, commercial viability, development time and cost calculations and the company’s commitment to improving patient care and outcomes today and in the future.
Multiple approaches to collaboration
To ensure that translational biology and scalable manufacturing processes advance in tandem to support commercially viable products, Pfizer engages with partners throughout the clinical, research, regulatory, academic and advocacy communities and with smaller, gene therapy-focused biotechnology companies. This is particularly true with respect to rare diseases, for which there are often just a handful of clinicians and researchers with sufficient relevant expertise and hands-on patient experience.
A recent example of the partnership-focused gene therapy ecosystem comes from Spark Therapeutics, Pfizer’s partner for hemophilia B gene therapy. Apart from Spark’s hemophilia B collaboration with Pfizer, their LUXTURNA gene therapy for confirmed biallelic RPE65 mutation-associated retinal dystrophy resulted from 25 years of research by Jean Bennett, PhD, and Al Maguire, MD, at Penn Medicine’s Center for Advanced Retinal and Ocular Therapeutics group in collaboration with the Children’s Hospital of Philadelphia (CHOP) in the United States (www.pennmedicine.org). The 2013 spinoff of Spark from CHOP enabled the company to develop its landmark gene therapy trial that led to its 2017 FDA approval (sparktx.com; www.blindness.org)7. Similarly, research from the network of academics and clinicians at the Nationwide Children’s Hospital’s Center for Gene Therapy in Columbus, Ohio in the US has led to the spinoff of several companies with gene therapies in clinical trials, including AveXis.
Seamless integration of technological expertise and resources across key communities should enable an informed development process that can reduce the time and cost needed to develop therapies that truly address patients’ clinical needs and quality of life concerns.
Consortia participation
Another way in which Pfizer collaborates with other key gene therapy communities is through participation in a variety of consortia. Pfizer has participated in many consortia, including public–private partnerships such the Innovative Medicines Initiative (IMI), a European Union initiative that seeks to address specific, yet widespread healthcare challenges by facilitating the sharing of knowledge and resources among cross-disciplinary partners. We believe there is a significant opportunity to leverage this collaborative model for gene therapy, such as through IMI’s 'Think Big' initiative for advanced therapy medicinal products (ATMPs). This collaborative approach encourages the development of solutions to challenges associated with this therapeutic paradigm.
Engagement with patient advocacy organizations and patient communities
Individuals with rare diseases and the advocacy organizations that represent them have expertise in the ‘lived experience’ of their disease. They are often experts in the science and biology of their diseases and are focused on building their capabilities to engage with the medical and pharmaceutical industry communities in the drug development process. They can provide invaluable insights about the burden of disease and the impact of current therapies on their lives. These insights are critical in establishing a true patient-centric approach that values benefit, meaningfulness and access. This is particularly true with emerging technologies such as gene therapy, where uncertainty is a variable that must also be factored into any risk–benefit equation and patient choice. Patient advocacy associations can be quite impactful in educating regulators about risk–benefit profiles that the patient community views as acceptable, motivating patients and providers to participate in clinical trials and enabling scientific advancement through collaborations with academia and industry.
Opportunities for collaboration with patient and advocacy groups may differ from one community to another based on the unmet need and priorities. For patient communities in which gene therapy is advancing through clinical development and for whom there is historical clinical research experience (for example, DMD), opportunities for collaboration with industry may focus on community education, community engagement to optimize development programs, and/or creating shared expectations on what gene therapy can and cannot achieve for disease management. Pfizer works closely with several advocacy groups within the DMD community on coalition-model efforts to solve for common challenges in drug development, including the Collaborative Trajectory Analysis Project (cTAP: http://ctap-duchenne.org), the Duchenne Regulatory Science Consortia (D-RSC: https://c-path.org/programs/d-rsc) and Project HERCULES (HEalth Research Collaboration United in Leading Evidence Synthesis: http://hercules.duchenneuk.org). Pfizer is also advancing with Parent Project Muscular Dystrophy (www.parentprojectmd.org) important patient preference research seeking to obtain quantitative evidence on patient and caregiver views on benefit and risk of emerging therapies including gene therapy.
For patient communities where gene therapy may be in earlier pre-clinical or even discovery phases, opportunities for collaboration with industry may focus more on transparency about the research agenda, optimizing access to scientific thought-leadership and data, and advancing the science through de-risking strategies, such as cost-sharing and consortia.
Harmonizing strategies for drug approval and market access
Pfizer has been a part of ongoing conversations with regulators and payors that we believe are essential for harmonizing approaches for drug approvals and market access. A clear regulatory pathway and a regulatory environment that values gene therapy as a potentially life-changing treatment modality are critical for the success of the field as a whole. Regulatory agencies, especially the FDA under commissioner Scott Gottlieb, have recognized these needs and have recently drafted guidance for pre-clinical, clinical and chemistry, manufacturing and controls expectations.
Following the issuance of the draft guidance, Gottlieb made clear the need for both consistency and flexibility in harmonizing gene therapy regulatory guidelines (www.fda.gov). Consistent and clear regulatory expectations will be critical for enabling gene therapy as a robust therapeutic class with commercially viable timelines and development costs rather than as a collection of one-off products that each require a new, expensive and time-consuming regulatory process. This is of particular importance for the development of gene therapies for small patient populations to ensure economic viability. And as regulatory agencies seek to exercise flexibility to facilitate gene therapy development, there also remains a lack of consistency in where to offer such flexibility, which creates a challenge for global development. Companies such as Pfizer that are pursuing a global plan for product development will be well positioned to help drive common experience and shared learning among regulatory agencies.
Pfizer’s aims for future innovation
After decades of development, Pfizer believes that gene therapy for single gene disorders is on the cusp of becoming a robust therapeutic modality in a variety of disease indications, and it is a major focus of our efforts in rare disease. Additional advances in vector engineering, disease modelling and both clinical and commercial-scale manufacturing are essential to ensure that the full potential of this approach is realized for as many patients as possible. We hope that our continued innovation and collaborations with academia, industry and patients will allow us, and others in the field, to transform gene therapy from an interesting scientific concept into a broad portfolio of commercial products that improve patient care and outcomes.
- World Federation of Hemophilia Report on the Annual Global Survey 2015 (2016). Available from: http://elearning.wfh.org/resource/annualglobal-survey-2015 [Accessed Oct 10 2018].
- Ferrari, F. K., Samulski, T., Shenk, T. & Samulski, R. J. J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. Virol. 70, 3227–3234 (1996).
- Fisher, K. J. et al. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J Virol. 70, 520–532 (1996).
- Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
- Arruda, A. & Samelson-Jones, B. J. Factor IX Padua: from biochemistry to gene therapy. Blood 128, SCI-9 (2016).
- George, L. A. et al. Spk-9001: adeno-associated virus mediated gene transfer for hemophilia B achieves sustained mean factor IX activity levels of > 30% without immunosuppression. Blood 128, 3 (2016).
- Jameson, J. L. Communication from the Dean: FDA approval of gene therapy for blindness. Perelman School of Medicine, Univ. Pennsylvania (2017). Available from: https://www.med.upenn.edu/ evpdeancommunications/2017-12-19.html [Accessed Oct 10 2018].
- Steinberg, M. H., Chui, D. H. K., Dover, G. J., Sebastiani, P. & Alsultan, A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 123, 481–485 (2013).
Authors:
Anna P. Tretiakova,
John E. Murphy,
Michael Binks,
Paul Mensah,
Joseph Rabinowitz,
Douglas M. McCarty,
Katherine Beaverson,
Molly MacLeod,
Gregory LaRosa &
Seng H. Cheng
